南开新闻网讯 近日,我校软件学院讲师曾婉雯团队在预测基因表达及解释非编码变异方面获重要进展,研究成果以“Reusability report: Compressing regulatory networks to vectors for interpreting gene expression and genetic variants”为题,发表在Nature子刊《自然•机器智能》(Nature Machine Intelligence)上,期刊影响因子15.508。
全基因组关联研究(GWAS)是确定复杂疾病风险基因/位点的有效研究策略,为复杂疾病的研究指明了方向,为实现个性化诊断、预后和治疗奠定了坚实基础,促进了人类遗传学和基因组学研究的发展。然而GWAS研究所得的大多数风险位点都位于非编码区,尚不清楚这些位点是否直接与疾病功能相关。而精准解释GWAS非编码区的位点是实现精准医疗的重要先决条件。
单核苷酸多态性(SNPs)主要是指在基因组水平上由单个核苷酸的变异(碱基的转换或颠换、插入或缺失)所引起的DNA序列多态性。它是人类可遗传变异中最常见的一种。大多数的功能性非编码SNPs可以通过干扰转录因子的结合和调控元件的功能来改变基因的表达,从而发挥其作用。值得注意的是,这些调控元件具有高度的细胞类型特异性,这提示SNPs的功能性也有细胞类型特异性。因此需要在正确的组织和区域背景下对每一种细胞类型中活跃的调节元件进行分类和功能揭示,结合其所形成的调控网络,进而帮助阐明常见神经退行性疾病分子发病机制中的基因风险位点功能的重要性。
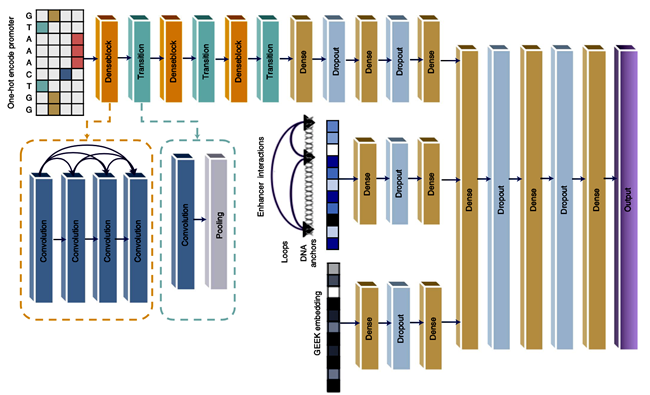
近年来,多组学技术尤其是表观染色质状态和三维结构测序技术得到了飞速发展与广泛应用,这对解析这些非编码区域变异的调控机制带来了新的契机。为系统解析调控网络对非编码遗传变异的影响,研究团队基于团队之前研发的DeepExpression模型,进一步整合序列数据、HiChIP三维基因组数据及GEEK模型调控网络数据的低维向量表示((low dimensional emedding),提高了对基因表达的预测能力。下游研究结果表明,整合序列数据、三维基因组数据和调控网络数据,能更好地理解转录调控机制和非编码变异的功能,为更精准解释GWAS遗传变异提供了新的方法。
南开大学软件学院为本文第一单位,南开大学软件学院讲师曾婉雯和斯坦福大学博士后信晶雪为共同第一作者,清华大学长聘副教授江瑞和中科院数学所王勇研究员为论文的共同通讯作者。该研究已得到国家自然科学基金青年项目资助、国家重点研发青年科学家项目资助。(软件学院供稿)
论文链接:https://www.nature.com/articles/s42256-021-00371-6
|


















